

I know, doctors and the media keep telling you cholesterol is evil. Why would you believe anybody else? You would do best to study the evidence for yourself:
Intake of up to 3 Eggs per Day Is Associated with Changes in HDL Function and Increased Plasma Antioxidants in Healthy, Young Adults
J. Nutr. 2017 147: 323
“Intake of 1 egg a day was sufficient to increase HDL function and large-LDL particle concentration; however, intake of 2–3 eggs/d supported greater improvements in HDL function as well as increased plasma carotenoids. Overall, intake of ≤3 eggs a day favored a less atherogenic LDL particle profile, improved HDL function, and increased plasma antioxidants in young, healthy adults.”
As reported before on this website, cholesterol is an essential molecule for our cell membranes, sex hormones, and neurons in the brain, among many other functions. It has been correctly implicated with plaque formation in our arteries, BUT, there is more to that story. Yes, cholesterol does up arteries that have become permeable due to inflammation and oxidation; this is another of its critical roles—maintaining the integrity and elasticity of arteries. This cannot is not done optimally when cholesterol is poorly managed in the liver. If our liver is “Fatty,”—an epidemic in this country—cholesterol is oxidized and inflamed in the liver. It becomes “sticky.” The inflamed and oxidized walls of arteries are also sticky. Think of Velcro. The result is poor patching up of arteries, which results in plaque formation. This is why plaques have cholesterol in them. But, don’t shoot the messenger. Surely you can see what the real problem is—processed foods, the main problem behind Fatty Liver.
The answer to cholesterol problem is not to demonize fats, except for artificial trans fats and industrially grown animals fattened beyond reason to maximize profits. The answer to stop eating refined foods high in sugar and trans fats and emphasize a plant-based diet. Then, Fatty Liver issues will be addressed, together with the epidemic of Pre Diabetes. These simple steps also address “leaky arteries.” Then, cholesterol will go down, grateful that it does not have to work so hard.
Statin drugs, you say? Sure, for some recalcitrant genetic cases. Most people only need to change their diet. Ponder this: statin drugs increase the risk of diabetes by fifty percent.
Here is a long article on the questionable cholesterol hypothesis:
Cholesterol Paradox: A Correlate Does Not a Surrogate Make
Evid Based Med. 2017;22(1):15-19.
The global campaign to lower cholesterol by diet and drugs has failed to thwart the developing pandemic of coronary heart disease around the world. Some experts believe this failure is due to the explosive rise in obesity and diabetes, but it is equally plausible that the cholesterol hypothesis, which posits that lowering cholesterol prevents cardiovascular disease, is incorrect. The recently presented ACCELERATE trial dumbfounded many experts by failing to demonstrate any cardiovascular benefit of evacetrapib despite dramatically lowering low-density lipoprotein cholesterol and raising high-density lipoprotein cholesterol in high-risk patients with coronary disease. This clinical trial adds to a growing volume of knowledge that challenges the validity of the cholesterol hypothesis and the utility of cholesterol as a surrogate end point. Inadvertently, the cholesterol hypothesis may have even contributed to this pandemic. This perspective critically reviews this evidence and our reluctance to acknowledge contradictory information.
Introduction
Nobel laureates Brown and Goldstein published an editorial in 1996 predicting that “Exploitation of recent breakthroughs … may well end coronary disease as a major public health problem early in the next century.”[1] They based their optimism largely on ‘proof of the cholesterol hypothesis’ which posits that lowering serum cholesterol reduces the risk of coronary heart disease (CHD). Paradoxically, CHD is now pandemic. Some may argue that this pandemic is secondary to the global explosion of obesity and diabetes, but it is equally plausible that the cholesterol hypothesis is incorrect. The results of the recently presented ACCELERATE trial may hold the key to understanding this paradox.[2]
The cholesterol hypothesis has been debated for years, but in light of recent clinical trial results, a reappraisal of the evidence is warranted. Cholesterol is an ostensibly ideal surrogate target: it is present in atherosclerotic plaque; cholesterol is an established risk factor for CHD; Mendelian randomisation studies suggest benefit from lifelong reduced cholesterol levels and cholesterol-lowering drug trials have reduced the risk of cardiovascular (CV) events. Consequently, it seemed impossible that the gold standard of modern medical research—a large, double-blind, randomised controlled trial (RCT)—could undermine, rather than confirm, this theory. Yet the ACCELERATE trial reported that evacetrapib, a novel cholesteryl ester transfer protein inhibitor, reduced low-density lipoprotein (LDL) cholesterol by 37%, raised high-density lipoprotein (HDL) cholesterol by 130%, but produced no discernible reduction in CV events or mortality in high-risk patients. I believe the ACCELERATE trial adds to the chorus that cholesterol is not a valid surrogate end point.
Rudolf Virchow first described the microscopy of the atherosclerotic plaque, but Nikolay Anichkov is credited with elucidating the central role of cholesterol in atherosclerosis. Ironically, cholesterol is also essential for life as a key component of cell membranes, steroid hormones and bile acids. The Framingham Heart Study further clarified the role of cholesterol as a major risk factor for CHD.[3] Ideally, a risk factor should help us distinguish those individuals who will develop a disease from those who will not. Figure 1 illustrates this concept and the original Framingham cholesterol data. The cholesterol levels of Framingham participants who did and did not develop CHD are remarkably similar except when the cholesterol level was extremely low (<150 mg/dL) or extremely high (>380 mg/dL). For the vast majority of patients, cholesterol levels do not help us differentiate those who will and will not develop CHD.
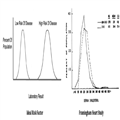
Figure 1.
Comparison of ideal risk factor with Framingham Heart Study cholesterol distribution in patients who developed coronary heart disease (CHD) and those that did not develop coronary heart disease (NON-CHD).3 Cholesterol values are mg/dL. Reprinted with permission of the publisher.
Mendelian randomisation studies are often cited in support of the cholesterol hypothesis. Conceptually, individuals born with genetically low LDL cholesterol should be protected from CHD since their cholesterol levels are reduced throughout life. Yet the report of PCSK9 sequence variations associated with low LDL cholesterol illustrates many of the shortcomings of this model.[4] This study reported that 2.6% of 3363 black patients in the Atherosclerosis Risk in Communities study had nonsense mutations in PCSK9 associated with a 28% reduction in LDL cholesterol. The authors calculated an 88% reduction in the risk of CHD by statistically comparing one fatal myocardial infarction in the PCSK9 group with 319 composite CHD events in the control group (unspecified, but defined as “definite or probable myocardial infarction, a silent myocardial infarction detected by electrocardiographic interval changes consistent with an intercurrent ischemic event, death due to CHD, or a coronary-revascularization procedure”). Such a comparison may not be valid and by ascribing equal importance to different events such as a CHD death and ischaemic electrocardiogram (EKG) changes the perceived benefit can easily be exaggerated.[5] Moreover, adjudicating CHD events based on death certificates and soft end points such as EKG changes limits the validity of the primary end point. Notably, this study reported no mortality or stroke benefit. These PCSK9 sequence variations were also associated with a statistically significant lower incidence of hypertension, which raises the question of whether LDL cholesterol lowering alone explains the reduction in CHD events. Medication and statin usage that might potentially impact CHD events were not reported. Ultimately, we must ask ourselves if this study proves the cholesterol hypothesis and should it be extrapolated to support the initiation of lipid lowering therapy in our adult patients? I believe Mendelian randomisation studies are hypothesis generating, not hypothesis proving.
Many experts cite numerous RCTs of statins in support of the cholesterol hypothesis, but we should not ignore the dozens of cholesterol-lowering trials that do not. Table 1 lists 44 cholesterol-lowering RCTs that reported no mortality benefit. Most reported no reduction in CV events, and several reported substantial harm (CDP, HERS, Minnesota Coronary Experiment, Sydney Diet Heart Study, WHI, WHO). This lack of benefit was seen even with profound reductions in LDL cholesterol (50% in the Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial). Although several studies were not specifically designed to assess mortality, the reported lack of mortality benefit should not be disregarded. While some experts have dismissed or criticised these negative trials, the totality of evidence simply cannot be ignored. Even when researchers demonstrate a statin mortality benefit, the findings are underwhelming. A recent analysis concluded that statins would only postpone death by a median of 3.1 and 4.2 days for primary and secondary prevention, respectively.[6]
Some researchers also point to meta-analyses as proof of the cholesterol hypothesis. Meta-analysis can provide an efficient mechanism of pooling similar, smaller studies and generating robust statistical results. But not all biostatisticians concur, and some refer to meta-analysis as ‘statistical alchemy for the twenty-first century’.[7] Moreover, the results of meta-analyses pertaining to cholesterol lowering are inconsistent. For example, the Cholesterol Treatment Trialists’ meta-analysis of 27 statin trials in people at low risk of vascular disease concluded that there was substantial benefit, but a subsequent meta-analysis of the same 27 statin trials concluded there was no mortality benefit.[8,9] Similarly, a meta-analysis of 11 statin trials in high-risk primary prevention found no mortality benefit and no correlation between the degree of LDL lowering and mortality rates.[10] Cochrane meta-analyses of cholesterol lowering in peripheral arterial disease of the lower extremities and statin use in acute coronary syndromes also reported no benefit.[11,12] Notably, the results of meta-analyses are often discordant with the results of subsequent large RCTs.[13]
Finally, consider that the cholesterol hypothesis may have inadvertently contributed to the very disease we seek to prevent. The cholesterol hypothesis risks oversimplifying the complex interaction of cholesterol, diet and coronary disease, leading many statin users to overeat with consequent obesity.[14] Nearly 50 years ago, three Harvard researchers were paid thousands of dollars by the sugar industry to write a review in the New England Journal of Medicine emphasising the importance of fat and cholesterol in CHD while minimising the importance of sugar.[15] Hence, the food industry developed and continues to promote low-cholesterol foods that are nonetheless high in sugar and refined carbohydrates. These dietary changes have likely contributed to the current epidemic of obesity and diabetes that can lead to CV disease.[16]
“A correlate does not a surrogate make,” and by definition, treatment of a valid surrogate end point should result in a consistent clinical benefit.[17] The empirical record is now clear that lowering cholesterol through diet or with eight different classes of drugs does not significantly prolong life or consistently prevent CHD (Table 1). Yet experts continue to proclaim the success of cholesterol lowering. Fifty-four years ago, Thomas Kuhn described this reluctance to acknowledge anomalies in a theory.[18] Dr Kuhn wrote that a paradigm shift would only occur when the evidence contradicting a theory is overwhelming. Therefore, we must accept the empirical record even though it contradicts our long-held beliefs. Other researchers believe this reluctance can be explained by the tendency to “see what you want to see,” and ignore what you do not.[19] For example, a recent editorial in the New England Journal of Medicine proclaimed, “Proof That Lower Is Better—LDL Cholesterol and IMPROVE-IT.”[20] IMPROVE-IT, a RCT of ezetimibe added to simvastatin in patients with a recent acute coronary syndrome, reported a 24% reduction in LDL cholesterol, but an absolute risk reduction in combined CV events of only 2% after 6 years. Furthermore, the results barely achieved statistical significance (HR 0.936, 95% CI 0.89 to 0.99) and there was no mortality benefit. The conclusions of this study must also be viewed cautiously since 42% of patients discontinued their study medications. The editorial further asserts that “IMPROVE-IT is a landmark study in that it is the first clinical trial to show a benefit of adding a nonstatin lipid-modifying agent to statin therapy.” Conspicuous by its absence is any mention of ENHANCE, another RCT of ezetimibe that reported no benefit when added to statin therapy in familial hypercholesterolaemic patients, or AIM-HIGH and HPS2-THRIVE, two RCTs that reported no benefit of niacin when added to statin therapy in patients with CV disease (Table 1).
theory.[18] Dr Kuhn wrote that a paradigm shift would only occur when the evidence contradicting a theory is overwhelming. Therefore, we must accept the empirical record even though it contradicts our long-held beliefs. Other researchers believe this reluctance can be explained by the tendency to “see what you want to see,” and ignore what you do not.[19] For example, a recent editorial in the New England Journal of Medicine proclaimed, “Proof That Lower Is Better—LDL Cholesterol and IMPROVE-IT.”[20] IMPROVE-IT, a RCT of ezetimibe added to simvastatin in patients with a recent acute coronary syndrome, reported a 24% reduction in LDL cholesterol, but an absolute risk reduction in combined CV events of only 2% after 6 years. Furthermore, the results barely achieved statistical significance (HR 0.936, 95% CI 0.89 to 0.99) and there was no mortality benefit. The conclusions of this study must also be viewed cautiously since 42% of patients discontinued their study medications. The editorial further asserts that “IMPROVE-IT is a landmark study in that it is the first clinical trial to show a benefit of adding a nonstatin lipid-modifying agent to statin therapy.” Conspicuous by its absence is any mention of ENHANCE, another RCT of ezetimibe that reported no benefit when added to statin therapy in familial hypercholesterolaemic patients, or AIM-HIGH and HPS2-THRIVE, two RCTs that reported no benefit of niacin when added to statin therapy in patients with CV disease (Table 1).
The debate over the cholesterol hypothesis has continued because the results of cholesterol lowering interventions are inconsistent and contradictory. Nevertheless, clinical guidelines continue to emphasize the critical importance of cholesterol lowering to prevent CHD. Unfortunately, I believe this one-dimensional approach may have impeded the advancement of science and our search for other preventive strategies. The ACCELERATE trial may well herald our tipping point and a sea change in our approach to CHD prevention.
The debate over the cholesterol hypothesis has continued because the results of cholesterol lowering interventions are inconsistent and contradictory. Nevertheless, clinical guidelines continue to emphasise the critical importance of cholesterol lowering to prevent CHD. Unfortunately, I believe this one-dimensional approach may have impeded the advancement of science and our search for other preventive strategies. The ACCELERATE trial may well herald our tipping point and a sea change in our approach to CHD prevention.
- References
- Brown MS, Goldstein JL. Heart attacks: gone with the century? Science 1996;272:629.
- Nicholls SJ, Lincoff A, Barter P, et al. Late-Breaking Clinical Trials II. The ACCELERATE trial: impact of the cholesteryl ester transfer protein inhibitor evacetrapib on cardiovascular outcome. Presented at the 65th Annual Scientific Session and Expo of the American College of Cardiology; April 2–4, Chicago, IL. 2016.
- Kannel WB, Castelli WP, Gordon T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann Intern Med 1979;90:85–91.
- Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264–72.
- Ferreira-González I, Busse JW, Heels-Ansdell D, et al. Problems with use of composite end points in cardiovascular trials: systemic review of randomized controlled trials. BMJ 2007;334:786.
- Kristensen ML, Christensen PM, Hallas J. The effect of statins on average survival in randomised trials, an analysis of end point postponement. BMJ Open 2015;5:e007118.
- Feinstein AR. Meta-analysis: statistical alchemy for the 21st century. J Clin Epidemiol 1995;48:71–9.
- Mihaylova B, Emberson J, Blackwell L, et al. Cholesterol Treatment Trialists’ (CTT) Collaborators. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomized trials. Lancet 2012;380:581–90.
- Abramson JD, Rosenberg HG, Jewell N, et al. Should people at low risk of cardiovascular disease take a statin? BMJ 2013;347:f6123.
- Ray KK, Seshasai SR, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med 2010;170:1024–31.
- Aung PP, Maxwell HG, Jepson RG, et al. Lipid lowering for peripheral arterial disease of the lower limb. Cochrane Database Syst Rev 2007;(4):CD000123.
- Vale N, Nordmann AJ, Schwartz GG, et al. Statins for acute coronary syndrome. Cochrane Database Syst Rev 2011;(6):CD006870.
- LeLorier J, Grégoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med 1997;337:536–42.
- Sugiyama T, Tsugawa Y, Tseng CH, et al. Different time trends of caloric and fat intake between statin users and nonusers among US adults: gluttony in the time of statins? JAMA Intern Med 2014;174:1038–45.
- Kearns CE, Schmidt LA, Glantz SA. Sugar industry and coronary heart disease research. A historical analysis of internal industry documents. JAMA Intern Med 2016;176:1680–5. Published online 12 September 2016.
- Gross LS, Li L, Ford ES, et al. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: an ecological assessment. Am J Clin Nutr 2004;79:774–9.
- Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med 1996;125:605–13.
- Kuhn TS. The structure of scientific revolutions. Chicago: University of Chicago Press, 1962.
- McCormack J, Greenhalgh T. Seeing what you want to see in randomised controlled trials: versions and perversions of UKPDS data. United Kingdom prospective diabetes study. BMJ 2000;320:1720–3.
- Jarcho JA, Keaney JF. Proof that lower is better—LDL cholesterol and IMPROVE-IT. N Engl J Med 2015;372:2448–50.